
Le concept de normes d’étiquetage médical souligne que le fonctionnement sûr d’un dispositif médical ne doit jamais dépendre de conjectures ou d’essais et d’erreurs..
Ces normes garantissent qu’un étiquetage informatif et précis n’est pas seulement une option mais une nécessité, chaque étiquette qui accompagne l’appareil devant être conforme aux normes et réglementations en vigueur.
Lorsque les caractéristiques ou les dangers ne sont pas correctement étiquetés, la porte est ouverte à une mauvaise utilisation, ce qui peut nuire à l’utilisateur et éroder la confiance dans la fiabilité de l’appareil. . Un étiquetage approprié contribue à prévenir les erreurs, garantissant que les utilisateurs obtiennent en toute sécurité les avantages escomptés de l’appareil.
L’étiquetage va au-delà d’une simple formalité dans le processus de production pour les fabricants de dispositifs médicaux ; c’est indispensable. Sans les étiquettes nécessaires, les appareils ne peuvent pas être mis sur le marché.
Les réglementations régissent tous les aspects de l’étiquetage, de la conception des étiquettes aux méthodes utilisées pour les apposer sur un appareil, obligeant les fabricants à accorder à l’étiquetage le même niveau d’importance qu’à toute autre facette de l’assurance qualité des produits..
Cette discussion vise à éclairer ce qui constitue une étiquette de dispositif médical, à délimiter les cas où un tel étiquetage est impératif et à approfondir les aspects critiques de la réglementation sur l’étiquetage des dispositifs médicaux aux États-Unis et dans l’Union européenne. .
Suite à cette exploration, nous proposerons des directives générales conçues pour aider les fabricants à créer des étiquettes qui profitent non seulement à leurs clients, mais qui sont également conformes aux exigences réglementaires, garantissant que leurs appareils sont utilisés de manière sûre et efficace. .
Qu’est-ce qui est exactement considéré comme un dispositif médical en termes juridiques ?
Légalement, la définition de la FDA englobe tout élément utilisé pour diagnostiquer, guérir, traiter, ou prévenir maladies ou problèmes de santé, ou qui modifie de manière significative la structure ou la fonction du corps. Cette vaste catégorie comprend diverses formes d’instruments, d’appareils, de machines, d’implants ou de réactifs in vitro, ainsi que toutes pièces, composants ou accessoires liés à ces appareils.
Notamment, les substances métabolisées par l’organisme, telles que les médicaments, ne relèvent pas de la catégorie ” appareil ” .. Il est intéressant de noter que les logiciels entrent également dans le domaine des dispositifs médicaux s’ils contribuent directement à l’une des fonctions susmentionnées, bien que les logiciels soient simplement utilisés pour stocker des données. les données ou les fins administratives sont exclues.
Dans le même ordre d’idées, le règlement sur les dispositifs médicaux de l’Union européenne identifie les dispositifs médicaux comme des produits non pharmacologiques conçus pour diagnostiquer, guérir et/ou prévenir des affections et des maladies. . Ces dispositifs peuvent également être utilisés pour étudier des processus anatomiques, physiologiques ou pathologiques. à des fins de remplacement et/ou de modification, ou pour collecter et conserver des échantillons de patients humains à des fins d’analyse médicale.
La question de savoir quelle partie de la chaîne d’approvisionnement d’un appareil est responsable de son étiquetage n’apporte pas toujours une réponse claire. . Généralement, la responsabilité incombe au fabricant de l’appareil, mais il existe des cas où d’autres parties impliquées peuvent également assumer des tâches d’étiquetage. . Cette liste peut être étendue. inclure:
- Retraiteurs d’appareils précédemment utilisés,
- Reconditionneurs d’appareils,
- Entités qui ré-étiquetent les appareils,
- Les assembleurs de kits compilent divers composants de l’appareil,
- Les développeurs de spécifications fournissent des descriptions détaillées des appareils,
- Et même les fabricants qui ne sont pas sûrs de leurs obligations spécifiques en matière d’étiquetage. .
Pour les fabricants aux prises avec des incertitudes concernant leurs responsabilités en matière d’étiquetage, il est conseillé de consulter les lignes directrices émises par leur organisme de réglementation. . Cette étape garantit la conformité et favorise une meilleure compréhension de leur rôle dans la protection de la sécurité des consommateurs grâce à un étiquetage approprié des appareils. .
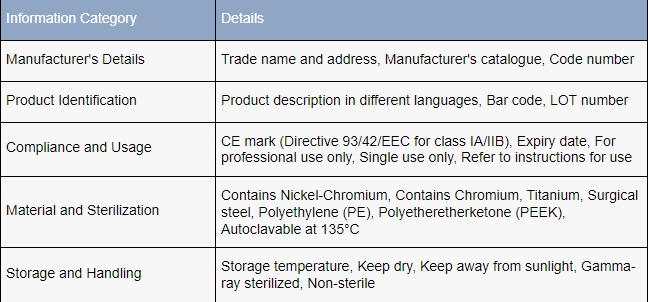
Que doivent savoir les fabricants sur la réglementation en matière d’étiquetage des dispositifs médicaux ?
Aux États-Unis et dans l’Union européenne, les exigences relatives à l’étiquetage des dispositifs médicaux sont méticuleusement détaillées et complètes, souvent adaptées à la catégorie spécifique du dispositif. .
Il est impératif que les fabricants consultent directement le libellé exact des lois et réglementations pertinentes, afin de s’assurer que leurs pratiques s’alignent précisément sur les exigences légales et restent conformes..
Cette diligence est cruciale non seulement pour le respect des normes légales, mais également pour la sécurité et l’utilisation éclairée des dispositifs médicaux par les professionnels de santé et les patients..
Le mandat de la FDA américaine pour les étiquettes des dispositifs médicaux
La Food and Drug Administration (FDA) des États-Unis définit une étiquette comme tout « affichage de contenu écrit, imprimé ou graphique sur le contenant immédiat de tout article », nécessitant une visibilité claire même lorsque le “conteneur immédiat” est en outre enfermé.
Le terme « étiquette » englobe en outre tout document imprimé sur un contenant ou un emballage de produit, ou tout élément accompagnant le produit au point de vente. Grâce à cette définition large, Les tribunaux américains ont déterminé que la publicité, les affiches, les brochures, les manuels d’instructions, les encarts et tout matériel analogue sont également classés comme étiquettes à des fins de surveillance réglementaire. .
Les exigences spécifiques en matière d’étiquetage sont énumérées dans le CFR Title 21, Part 801, qui stipule que les étiquettes conformes à la FDA doivent comporter :
- Le nom et l’adresse professionnelle du fabricant,
- L’application prévue de l’appareil,
- Des instructions complètes qui permettent à un profane d’utiliser l’appareil en toute sécurité sans l’aide d’un professionnel.
Les étiquettes doivent éviter toute affirmation fausse ou trompeuse et être placées bien en vue à un endroit approprié sur l’emballage.
De plus, la FDA oblige les fabricants à incorporer des étiquettes d’identification unique des appareils (UDI) pour faciliter le suivi précis des appareils.. Ces UDI doivent être lisibles par les humains et les machines, garantissant que les éléments de suivi répondent à des normes rigoureuses de clarté et d’accessibilité..

Les prescriptions d’étiquetage selon le MDR de l’UE
Le Règlement sur les dispositifs médicaux (MDR) de l’Union européenne englobe ses réglementations en matière d’étiquetage dans l’Annexe I, Chapitre III. Ces réglementations correspondent globalement à celles de la FDA, y compris le mandat d’identification unique des dispositifs (UDI) sur les étiquettes.
Au-delà de l’apposition d’étiquettes directement sur le dispositif médical ou son emballage, le cas échéant, Les fabricants de l’UE sont également tenus de fournir des informations d’étiquetage à jour via leur site Web d’entreprise.. Cette exigence garantit la transparence et l’accessibilité des informations, permettant aux utilisateurs finaux et aux organismes de réglementation d’accéder et de consulter facilement les détails d’étiquetage pertinents pour les dispositifs médicaux..
Pourquoi faut-il accorder une importance primordiale à l’étiquetage des dispositifs médicaux ?
Un étiquetage clair et précis sur les dispositifs médicaux revêt une importance cruciale, d’autant plus que nombre de ces dispositifs sont destinés à être utilisés par les patients à domicile, sans la supervision de professionnels de santé..
Pour garantir la sécurité et l’efficacité, non seulement les patients, mais aussi les professionnels de la santé et les utilisateurs chevronnés s’appuient largement sur des instructions, des conseils et des mises en garde claires..
En cas de mauvaise utilisation ou de manipulation incorrecte, les répercussions pour l’utilisateur peuvent aller du dysfonctionnement de l’appareil à des blessures graves, voire la mort.. Même des faux pas mineurs peuvent avoir des effets néfastes sur la santé du patient..
Pour chaque partie impliquée dans le cycle de vie de l’appareil, de la conception à la livraison, le bien-être de l’utilisateur final doit être la priorité absolue..
De bonnes pratiques d’étiquetage constituent un outil essentiel pour réduire les incidents indésirables et maintenir la confiance dans les dispositifs médicaux. . De plus, ces pratiques ne visent pas seulement à protéger la santé des utilisateurs ; ils concernent également le respect des normes réglementaires.
Un étiquetage efficace peut éviter les inconvénients liés à la non-conformité, tels que des retards coûteux, des rappels ou des problèmes juridiques, garantissant ainsi une livraison ininterrompue des dispositifs médicaux sur le marché.
Les principes clés de la création d’étiquettes de dispositifs médicaux
Construire une étiquette de dispositif médical exemplaire n’est pas la recherche d’un modèle universel mais un processus nuancé. Les éléments cruciaux varient considérablement en fonction d’une myriade de facteurs, il incombe aux fabricants de garantir l’inclusion de tous les composants vitaux.
L’objectif d’une étiquette n’est pas seulement de remplir les mandats réglementaires, mais de garantir qu’elle transmet toutes les informations nécessaires pour que le patient puisse tirer un résultat positif de l’utilisation du dispositif. .
Pour atteindre un tel objectif, voici un aperçu détaillé des bonnes pratiques pour le développement de labels efficaces et conformes :
- Initier les considérations de conception d’étiquettes dès les premières étapes du développement du produit, pour intégrer l’étiquetage de manière transparente à la conception et à la fonction de l’appareil. .
- Consultez avec diligence les directives spécifiques établies par votre organisme de réglementation pour vérifier l’étendue complète des informations dont l’inclusion est obligatoire, en particulier dans le mode d’emploi..
- Utiliser des symboles standardisés conformes aux réglementations internationales pour garantir une compréhension et une utilisation universelles.
- Optez pour des matériaux d’étiquettes robustes, capables de résister à des processus de nettoyage et de stérilisation répétés tout en conservant la lisibilité. .
- Rationalisez la production d’étiquettes grâce à l’automatisation pour améliorer la cohérence et réduire le risque d’erreur humaine.
- Créez des modèles d’étiquettes adaptables qui sont facilement mis à jour avec les informations nécessaires pour accélérer le processus d’étiquetage de divers produits.
- Collaborer étroitement avec les partenaires de la chaîne d’approvisionnement pour synchroniser les efforts de conformité et éviter les tâches d’étiquetage redondantes.
- Effectuez des tests d’utilisabilité approfondis pour confirmer que le message prévu de vos étiquettes est interprété et compris avec précision par les utilisateurs.
- Incorporer des avertissements complets qui détaillent tous les risques connus et indiquent clairement les implications d’une utilisation non conforme.
- Avant de consolider votre approche d’étiquetage, effectuez un examen approfondi du cadre juridique pour déterminer toute exigence d’étiquetage supplémentaire et spécialisée pertinente à vos dispositifs médicaux spécifiques. . Cette étape garantit que vos étiquettes sont non seulement informatives, mais également conformes à la loi, en conformité avec les réglementations et normes.
QMS et étiquetage des dispositifs médicaux
Un système de gestion de la qualité (QMS) méticuleusement adapté aux exigences uniques des fabricants de dispositifs médicaux constitue un élément essentiele, garantissant le respect des exigences réglementaires et soutenant la production d’étiquettes qui reflètent la haute qualité des appareils qu’elles accompagnent.
Une solution QMS efficace renforcera les protocoles d’étiquetage en conservant les données de fabrication à jour, en suivant méticuleusement les révisions des documents et en vérifiant que chaque aspect du processus d’étiquetage est en stricte conformité avec toutes les réglementations pertinentes.
De plus, le déploiement d’un système de gestion de la qualité joue un rôle déterminant dans la création d’une approche standardisée de l’étiquetage, où la cohérence de la qualité des étiquettes devient aussi critique que la qualité de l’appareil. . Le système aide à identifier les non-conformités potentielles avant qu’elles ne deviennent des problèmes, améliorant ainsi l’efficacité opérationnelle globale. .
En intégrant un système de gestion de la qualité robuste, les fabricants peuvent être assurés que leurs étiquettes sont exactes, à jour et entièrement alignées sur les normes établies par l’industrie, renforçant ainsi la confiance et la sécurité des prestataires de soins de santé et des patients..
Orientations futures possibles pour l’étiquetage des dispositifs médicaux
À mesure que la technologie progresse et que les exigences du secteur de la santé évoluent, l’avenir de l’étiquetage des dispositifs médicaux devrait mettre davantage l’accent sur les technologies intelligentes, la personnalisation et la durabilité environnementale.. Voici quelques tendances anticipées :
- Intégration des technologies d’étiquettes intelligentes: Les futures étiquettes de dispositifs médicaux pourraient intégrer des technologies avancées telles que NFC (Near Field Communication), RFID (Radio Frequency Identification) et des capteurs.. Ces technologies pourraient permettre un suivi en temps réel de l’utilisation et de l’emplacement des appareils, améliorant ainsi l’efficacité et la précision de la gestion des appareils..
- Personnalisation et personnalisation accrues: Grâce aux progrès des technologies d’impression, telles que l’impression 3D, les étiquettes de dispositifs médicaux pourraient atteindre des niveaux plus élevés de personnalisation et de personnalisation. Les prestataires de soins de santé pourraient adapter le contenu des étiquettes pour répondre à des besoins cliniques spécifiques, mieux servir les patients et les professionnels de la santé.
- Utilisation de matériaux écologiques: En réponse aux préoccupations environnementales croissantes, les futurs labels utiliseront probablement des matériaux recyclables ou biodégradables, réduisant ainsi l’impact environnemental et s’alignant sur les efforts mondiaux de développement durable.
- Informations complètes sur la conformité réglementaire et la sécurité: À mesure que les réglementations internationales deviennent plus strictes, les étiquettes des dispositifs médicaux devront inclure des instructions de sécurité et des déclarations de conformité plus détaillées. Cela aidera les prestataires de soins de santé à mieux comprendre et utiliser les dispositifs tout en garantissant la sécurité des patients..
- Interaction améliorée avec les patients: Les étiquettes peuvent inclure des éléments interactifs tels que des codes QR renvoyant vers des vidéos pédagogiques, des manuels d’utilisation et d’autres ressources éducatives pour les patients. . Cette interaction peut encourager les patients à jouer un rôle actif dans leur traitement, améliorant ainsi l’observance et les résultats. .
Conclusion
Bref, le respect de normes d’étiquetage médical est essentiel pour garantir la sécurité des patients et maintenir la confiance dans les dispositifs médicaux, et les étiqueteuses jouent un rôle central dans ce .
Nous sommes une entreprise qui fabrique des étiqueteuses pharmaceutiques polyvalentes conçues pour répondre à ces normes pour une large gamme d’applications de dispositifs médicaux, notamment flacons, seringues, et ampoules. Pour les fabricants recherchant fiabilité et conformité dans le processus d’étiquetage, nous sommes votre partenaire fiable !